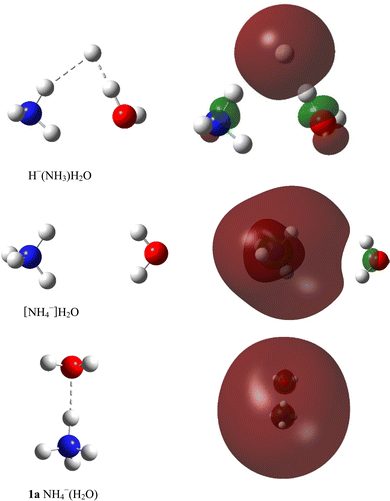
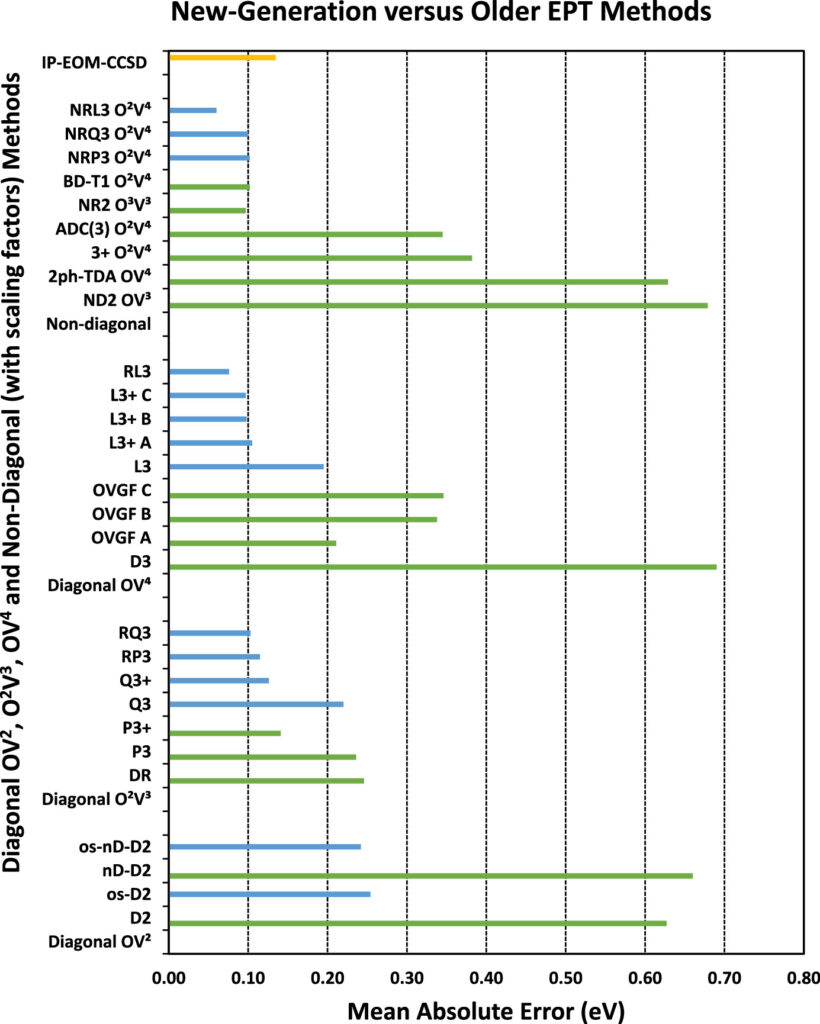
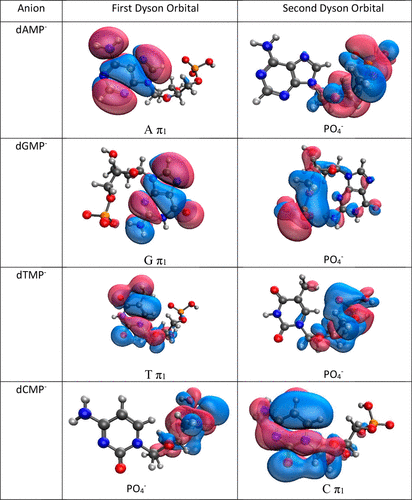
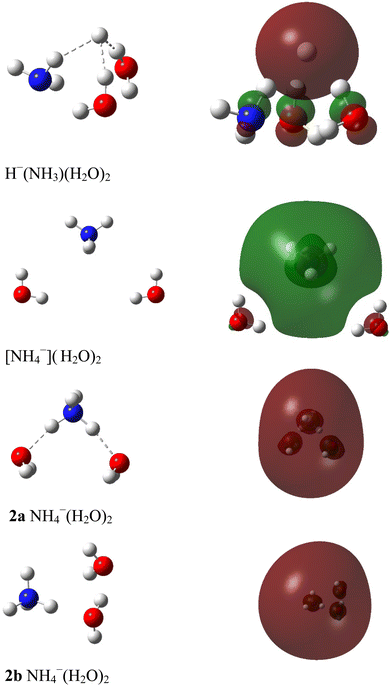
Molecular quantum chemistry is a branch of physical chemistry focused on the application of quantum mechanics to chemical systems, particularly towards the quantum-mechanical calculation of electronic contributions to physical and chemical properties of molecules, materials, and solutions at the atomic level. These calculations include systematically applied approximations intended to make calculations computationally feasible while still capturing as much information about important contributions to the computed wave functions as well as to observable properties such as structures, spectra, and thermodynamic properties. Quantum chemistry is also concerned with the computation of quantum effects on molecular dynamics and chemical kinetics.
Chemists rely heavily on spectroscopy through which information regarding the quantization of energy on a molecular scale can be obtained. Common methods are infra-red (IR) spectroscopy, nuclear magnetic resonance (NMR) spectroscopy, and scanning probe microscopy. Quantum chemistry may be applied to the prediction and verification of spectroscopic data as well as other experimental data.
Many quantum chemistry studies are focused on the electronic ground state and excited states of individual atoms and molecules as well as the study of reaction pathways and transition states that occur during chemical reactions. Spectroscopic properties may also be predicted. Typically, such studies assume the electronic wave function is adiabatically parameterized by the nuclear positions (i.e., the Born–Oppenheimer approximation). A wide variety of approaches are used, including semi-empirical methods, density functional theory, Hartree-Fock calculations, quantum Monte Carlo methods, coupled cluster methods, and configuration interaction methods.
Understanding electronic structure and molecular dynamics through the development of computational solutions to the Schrödinger equation is a central goal of quantum chemistry. Progress in the field depends on overcoming several challenges, including the need to increase the accuracy of the results for small molecular systems, and to also increase the size of large molecules that can be realistically subjected to computation, which is limited by scaling considerations — the computation time increases as a power of the number of atoms.
The primary goal of our department is to develop and apply new and existing quantum mechanical methodologies to study wide array of chemical phenomena: molecular spectroscopy, ground and excited state properties, reaction mechanisms, computational catalysis, prediction of new exotic molecules and ions, development of direct quantum chemistry methods etc.
Collaborations with theory and experimental groups are always welcome, especially, those of African natives. Reach us via: info@nesvard.org